第 36 章 模型比较和拟合优度
我们用数据拟合广义线性模型时其实有许多不同的目的和意义:
- 估计某些因素的暴露和因变量之间的相关程度,同时调整其余的混杂因素;
- 确定能够强有力的预测因变量变化的因子;
- 用于预测未来的事件或者病人的预后等等。
但是一般情况下,我们拿到数据以后不可能立刻就能构建起来一个完美无缺的模型。我们常常会拟合两三个甚至许多个模型,探索模型和数据的拟合程度,就成为了比较哪个模型更优于其他模型的硬指标。本章的目的是介绍 GLM 嵌套式模型之间的两两比较方法,其中一个模型的预测变量是另一个模型的预测变量的子集。
对手里的数据构建一个GLM的过程,其实就是在该数据的条件下(given the data),对模型参数\(\mathbf{\beta}\) 定义其对数似然(log-likelihood),并寻找能给出极大值的那一系列极大似然估计(maximum likelihood estimates, MLE) \(\mathbf{\hat\beta}\) 的过程。每次构建一个模型,我们都会获得该模型对应的极大对数似然,它其实是极为依赖构建它的观察数据的,意味着每次观察数据发生变化,你即使用了相同的模型来拟合相同的GLM获得的极大似然都会发生变化。所以其实我们并不会十分关心这个极大似然的绝对值大小。我们关心的其实是,当对相同数据,构建了包含不同变量的模型时,极大似然的变化量。因为这个极大似然(或者常被略称为对数似然 log likelihood,甚至直接只叫做似然)的变化量本身确实会反应我们思考的模型,和观察数据之间的拟合程度。一般来说,模型中变量较少的那个(通常叫做更加一般化的模型more general model)获得的似然值和变量较多的那个模型获得的似然值相比较都会比较小,我们关心的似然值在增加了新变量之后的复杂模型后获得的增量,是否有价值,是否真的改善了模型的拟合度(whether the difference in log likelihoods is large enough to indicate that the less general model provides a “real” improvement in fit)。
36.1 嵌套式模型的比较 nested models
假如我们用相同的数据拟合两个 GLM,\(\text{Model 1, Model 2}\)。其中,当限制\(\text{Model 2}\) 中部分参数为零之后会变成\(\text{Model 1}\)时, 我们说\(\text{Model 1}\) 是\(\text{Model 2}\) 的嵌套模型。
- 例1:嵌套式模型 I
模型 1 的线性预测方程为 \[\eta_i = \alpha + \beta_1 x_{i1}\]
模型2 和模型1 的因变量相同(分布相同),使用相同的链接方程(link function) 和尺度参数(scale parameter, \(\phi\)),但是它的线性预测方程为\[\eta_i = \alpha + \beta_1 x_{i1} + \beta_2 x_{i1} + \beta_3 x_{i3}\]
此时我们说模型 1 是模型 2 的嵌套模型,因为令 \(\beta_2 = \beta_3 = 0\) 时,模型 2 就变成了 模型 1。 - 例2:嵌套式模型 II
模型 1 的线性预测方程为 (此处默认 \(x_{i1}\) 是连续型预测变量) \[\eta_i = \alpha + \beta_1 x_{i1}\]
模型 2 的线性预测方程如果是 \[\eta_i = \alpha + \beta_1 x_{i1} + \beta_2 x^2_{i1}\]
此时我们依然认为 模型 1 是模型 2 的嵌套模型, 因为令 \(\beta_2 = 0\) 时,模型 2 就变成了 模型 1。
关于嵌套式模型,更加一般性的定义是这样的:标记模型2 的参数向量是\(\mathbf{(\psi, \lambda)}\),其中,当我们限制了参数向量的一部分例如$ $,模型2 就变成了模型1 的话,模型1 就是嵌套于模型2 的。所以比较嵌套模型之间的拟合度,我们可以比较较为复杂的 模型 2 相较 模型 1 多出来的复杂的预测变量参数部分 \(\mathbf{\psi}\) 是否是必要的。也就是说,比较嵌套模型哪个更优的情况下,零假设是 \(\mathbf{\psi = 0}\)。
这是典型的多变量的模型比较,需要用到子集似然比检验 ??,log-likelihood ratio test:
\[ \begin{aligned} -2pllr(\psi = 0) & = -2\{ \ell_p(\psi=0) - \ell_p(\hat\psi) \} \stackrel{\cdot}{\sim} \chi^2_{df} \\ \text{Where } \hat\psi & \text{ denotes the MLE of } \psi \text{ in Model 2} \\ \text{With } df & = \text{ the dimension of } \mathbf{\psi} \end{aligned} \]
\(\ell_p(\psi=0)\),其实是 模型 1 的极大对数似然,记为 \(\ell_1\)。 \(\ell_p(\hat\psi)\) 其实是 模型 2 的极大对数似然,记为 \(\ell_2\)。所以这个似然比检验统计量就变成了:
\[ -2pllr(\psi = 0) = -2(\ell_1-\ell_2) \]
这个统计量在零假设的条件下服从自由度为两个模型参数数量之差的卡方分布。如果 \(p\) 值小于提前定义好的显著性水平,将会提示有足够证据证明 模型 2 比 模型 1 更好地拟合数据。
36.2 嵌套式模型比较实例
回到之前用过的疯牛病和牲畜群的数据 35.4。我们当时成功拟合了两个 GLM 模型,模型 1 的预测变量只有 “饲料”,“群”;模型 2 的预测变量在模型 1 的基础上增加二者的交互作用项。并且我们当时发现交互作用项部分并无实际统计学意义 \(p = 0.584\)。现在用对数似然比检验来进行类似的假设检验。
先用logLik(Model) 的方式提取两个模型各自的对数似然,然后计算对数似然比,再去和自由度为1 (因为两个模型只差了1 个预测变量) 的卡方分布做比较:
Model1 <- glm(cbind(infect, cattle - infect) ~ factor(group) + dfactor, family = binomial(link = logit), data = Cattle)
Model2 <- glm(cbind(infect, cattle - infect) ~ factor(group) + dfactor + factor(group)*dfactor, family = binomial(link = logit), data = Cattle)
logLik(Model1)## 'log Lik.' -13.12687 (df=3)## 'log Lik.' -12.97384 (df=4)LLR <- -2*(logLik(Model1) - logLik(Model2))
1-pchisq(as.numeric(LLR), df=1) # p value for the LLR test## [1] 0.5801037再和 lmtest::lrtest 的输出结果作比较。
## Likelihood ratio test
##
## Model 1: cbind(infect, cattle - infect) ~ factor(group) + dfactor
## Model 2: cbind(infect, cattle - infect) ~ factor(group) + dfactor + factor(group) *
## dfactor
## #Df LogLik Df Chisq Pr(>Chisq)
## 1 3 -13.127
## 2 4 -12.974 1 0.3061 0.5801结果跟我们手计算的结果完全吻合。 AWESOME !!!
值得注意的是,此时进行的似然比检验结果获得的p 值,和模型中Wald 检验结果获得的p 值十分接近(0.5801 v.s. 0.584),这也充分显示了这两个检验方法其实是渐进相同的(asymptotically equivalent)。
36.3 饱和模型,模型的偏差,拟合优度
在简单线性回归中,残差平方和提供了模型拟合数据好坏的指标– 决定系数\(R^2\) (Section 25.2.3),并且在偏F 检验(Section @ref( partialF)) 中得到模型比较的应用。
广义线性回归模型中事情虽然没有这么简单,但是思想可以借鉴。先介绍饱和模型 (saturated model) 的概念,再介绍其用于模型偏差 (deviance) 比较的方法。前文中介绍过的嵌套模型之间的对数似然比检验,也是测量两个模型之间偏差大小的方法。
36.3.1 饱和模型 saturated model
饱和模型 saturated model,是指一个模型中所有可能放入的参数都被放进去的时候,模型达到饱和,自由度为零。其实就是模型中参数的数量和观测值个数相等的情况。饱和模型的情况下,所有的拟合值和对应的观测值相等。所以,对于给定的数据库,饱和模型提供了所有模型中最“完美” 的拟合值,因为拟合值和观测值完全一致,所以饱和模型的对数似然,比其他所有你建立的模型的对数似然都要大。但是多数情况下,饱和模型并不是合理的模型,不能用来预测也无法拿来解释数据,因为它本身就是数据。
36.3.2 模型偏差 deviance
令 \(L_c\) 是目前拟合模型的对数似然,\(L_s\) 是数据的饱和模型的对数似然,所以两个模型的对数似然比是 \(\frac{L_c}{L_s}\)。那么尺度化的模型偏差 (scaled deviance) \(S\) 被定义为:
\[ S=-2\text{ln}(\frac{L_c}{L_s}) = -2(\ell_c - \ell_s) \]
值得注意的是,非尺度化偏差(unscaled deviance) 被定义为\(\phi S\),其中的\(\phi\) 是尺度参数,由于泊松分布和二项分布的尺度参数都等于1 (\(\phi = 1\)),所以尺度化偏差和非尺度化偏差才会在数值上相等。
这里定义的模型偏差大小,可以反应一个模型拟合数据的程度,偏差越大,该模型对数据的拟合越差。 “Deviance can be interpreted as Badness of fit”.
但是,模型偏差只适用于汇总后的二项分布数据(aggregated)。当数据是个人的二进制数据时 (inidividual binary data),模型的偏差值变得不再适用,无法用来比较模型对数据的拟合程度。 这是因为当你的观测值 (个人数据) 有很多时,拟合饱和模型所需要的参数个数会趋向于无穷大,这违背了子集对数似然比检验的条件。
36.3.3 汇总型二项分布数据 aggregated/grouped binary data
假如,观察数据是互相独立的,服从二项分布的 \(n\) 个观测值: \(Y_i \sim Bin(n_i, \pi_i), i=1,\dots,n\)。用汇总型的数据表达方法来描述它,那么获得的数据就是一个个分类变量在各自组中的人数或者百分比的数据 (如下面的数据所示)。这样的数据的饱和模型,其实允许了每个分类变量的组中百分比变化 (The saturated model for this data allows the probability of “success” to be different in each group,so that \(\tilde{\pi} = \frac{y_i}{n_i}\))。也就是每组的模型拟合后百分比,等于观察到的百分比。
## dose n_deaths n_subjects
## 1 49.06 6 59
## 2 52.99 13 60
## 3 56.91 18 62
## 4 60.84 28 56
## 5 64.76 52 63
## 6 68.69 53 59
## 7 72.61 60 62
## 8 76.54 59 60那么汇总型二项分布数据,其饱和模型的对数似然其实就是
\[ \ell_s = \sum_{i = 1}^n\{ \log\binom{n_i}{y_i} + y_i\log(\tilde{\pi_i}) + (n_i - y_i)\log(1 - \tilde{ \pi_i}) \} \]
假设此时我们给这个数据拟合一个非饱和模型,该模型告诉我们每个分类组中的预测百分比是\(\hat\pi_i, i = 1, \dots, n\),那么这个非饱和模型的对数似然其实是
\[ \ell_c = \sum_{i = 1}^n\{ \binom{n_i}{y_i} + y_i\log(\hat\pi_i) + (n_i - y_i)\log(1-\hat\pi_i)\} \]
那么这个非饱和模型的模型偏差 (deviance) 就等于
\[ \begin{aligned} S & = -2(\ell_c - \ell_s) \\ & = 2\sum_{i = 1}^n\{ y_i\log(\frac{\tilde{\pi_i}}{\hat\pi_i}) + (n_i - y_i)\log(\frac{1-\ tilde{\pi_i}}{1-\hat\pi_i}) \} \end{aligned} \]
从上面这个表达式不难看出,模型偏差值的大小,将会随着模型预测值的变化而变化,如果它更加接近饱和模型的预测值(饱和模型的预测值其实就等于观测值),那么模型的偏差就会比较小。如果你的汇总型数据拟合了你认为合适的模型以后,你发现它的模型偏差值很大,那么就意味着你的模型预测值其实和观测值相去甚远,模型和观测值的拟合度应该不理想。对于汇总型数据来说,模型偏差值,其实等价于将你拟合的模型和饱和模型之间做子集对数似然比检验 (profile log-likelihood ratio test)。渐进来说(asymptotically),这个子集对数似然比检验的结果,会服从自由度为\(n-p\) 的\(\chi^2\) 分布,其中\(n, p\) 分别是饱和模型和你拟合的模型中被估计参数的个数。
36.4 个人数据拟合模型的优度检验
在上文中已经提到了,当你的数据不再是汇总型二项分布数据,而是个人二项分布数据 (individual binary data) 时,模型偏差 (deviance) 无法用来评价你建立的模型。这样的数据其实比汇总型二项分布数据更加常见,当模型中一旦需要加入一个连续型变量时,数据就只能被表达为个人二项分布数据。对于个人二项分布数据模型拟合度比较,最常用的方法是 (Hosmer and Lemesbow 1980) 提出的模型拟合优度检验法 (goodness of fit)。该方法的主要思想是,把个人二项分布数据模型获得的个人预测值(model predicted probabilities) \(\hat\pi_i\) 进行人为的分组,把预测值数据强行变成汇总型二项分布数据,那么观测值的样本量即使增加到无穷大,也不会使得模型中组别增加到无穷大,从而可以规避
在 R 里面,进行逻辑回归模型的拟合优度检验的自定义方程如下:
hosmer <- function(y, fv, groups=10, table=TRUE, type=2) {
# A simple implementation of the Hosmer-Lemeshow test
q <- quantile(fv, seq(0,1,1/groups), type=type)
fv.g <- cut(fv, breaks=q, include.lowest=TRUE)
obs <- xtabs( ~ fv.g + y)
fit <- cbind( e.0 = tapply(1-fv, fv.g, sum), e.1 = tapply(fv, fv.g, sum))
if(table) print(cbind(obs,fit))
chi2 <- sum((obs-fit)^2/fit)
pval <- pchisq(chi2, groups-2, lower.tail=FALSE)
data.frame(test="Hosmer-Lemeshow",groups=groups,chi.sq=chi2,pvalue=pval)
}library(haven)
# lbw <- read_dta("http://www.stata-press.com/data/r12/lbw.dta")
lbw <- read_dta(file = "../backupfiles/lbw.dta")
lbw$race <- factor(lbw$race)
lbw$smoke <- factor(lbw$smoke)
lbw$ht <- factor(lbw$ht)
Modelgof <- glm(low ~ age + lwt + race + smoke + ptl + ht + ui,
data = lbw, family = binomial(link = logit))
hosmer(lbw$low, fitted(Modelgof))## 0 1 e.0 e.1
## [0.0273,0.0827] 19 0 17.822223 1.177777
## (0.0827,0.128] 17 2 16.973902 2.026098
## (0.128,0.201] 13 6 15.828544 3.171456
## (0.201,0.243] 18 1 14.695710 4.304290
## (0.243,0.279] 12 7 14.106205 4.893795
## (0.279,0.314] 12 7 13.360124 5.639876
## (0.314,0.387] 13 6 12.462805 6.537195
## (0.387,0.483] 12 7 10.824166 8.175834
## (0.483,0.594] 9 10 8.690142 10.309858
## (0.594,0.839] 5 13 5.236180 12.763820## test groups chi.sq pvalue
## 1 Hosmer-Lemeshow 10 9.650683 0.2904041## 0 1 e.0 e.1
## [0.0273,0.128] 36 2 34.79612 3.203876
## (0.128,0.243] 31 7 30.52425 7.475746
## (0.243,0.314] 24 14 27.46633 10.533671
## (0.314,0.483] 25 13 23.28697 14.713029
## (0.483,0.839] 14 23 13.92632 23.073679## test groups chi.sq pvalue
## 1 Hosmer-Lemeshow 5 2.435921 0.48698336.5 GLM例4
36.5.1 回到之前的昆虫数据,尝试评价该模型的拟合优度。
- 重新读入昆虫数据,拟合前一个练习中拟合过的模型,使用
glm()命令。
Insect <- read.table("../backupfiles/INSECT.RAW", header = FALSE, sep ="", col.names = c("dose", "n_deaths", "n_subjects"))
# print(Insect)
Insect <- Insect %>%
mutate(p = n_deaths/n_subjects)
Model1 <- glm(cbind(n_deaths, n_subjects - n_deaths) ~ dose,
family = binomial(link = logit), data = Insect)
summary(Model1); jtools::summ(Model1, digits = 6, confint = TRUE, exp = TRUE)##
## Call:
## glm(formula = cbind(n_deaths, n_subjects - n_deaths) ~ dose,
## family = binomial(link = logit), data = Insect)
##
## Coefficients:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -14.0864 1.2284 -11.47 <2e-16 ***
## dose 0.2366 0.0203 11.65 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## (Dispersion parameter for binomial family taken to be 1)
##
## Null deviance: 268.2683 on 7 degrees of freedom
## Residual deviance: 4.6155 on 6 degrees of freedom
## AIC: 37.394
##
## Number of Fisher Scoring iterations: 4| Observations | 8 |
| Dependent variable | cbind(n_deaths, n_subjects - n_deaths) |
| Type | Generalized linear model |
| Family | binomial |
| Link | logit |
| χ²(1) | 263.652801 |
| Pseudo-R² (Cragg-Uhler) | 1.000000 |
| Pseudo-R² (McFadden) | 0.887580 |
| AIC | 37.393985 |
| BIC | 37.552868 |
| exp(Est.) | 2.5% | 97.5% | z val. | p | |
|---|---|---|---|---|---|
| (Intercept) | 0.000001 | 0.000000 | 0.000008 | -11.467341 | 0.000000 |
| dose | 1.266925 | 1.217500 | 1.318357 | 11.653009 | 0.000000 |
| Standard errors: MLE |
- 根据上面模型输出的结果,检验是否有证据证明该模型对数据的拟合不佳。
上面模型拟合的输出结果中,可以找到最下面的模型偏差值的大小和相应的自由度: Residual deviance: 4.6155 on 6 degrees of freedom。如果我们要检验该模型中假设的前提条件之一–昆虫死亡的对数比值(on a log-odds scale) 和药物浓度(dose) 之间是线性关系(或者你也可以说,检验是否有证据证明该模型对数据拟合不佳),我们可以比较计算获得的模型偏差值在自由度为6 的卡方分布(\(\chi^2_6\)) 中出现的概率。这里自由度 6 是由 \(n - p = 8 - 2\) 计算获得,其中 \(n\) 是数据中观察值个数,\(p\) 是模型中估计的参数的个数。检验方法很简单:
## [1] 0.5939847所以,检验的结果,P 值就是 0.594,没有任何证据反对零假设(模型拟合数据合理)。
- 试比较两个模型对数据的拟合效果孰优孰劣:模型1,上面的模型;模型2,加入剂量的平方 (dose2),作为新增的模型解释变量。嵌套式模型之间的比较使用的是似然比检验法 (profile likelihood ratio test),试着解释这个比较方法和 Wald 检验之间的区别。
Model2 <- glm(cbind(n_deaths, n_subjects - n_deaths) ~ dose + I(dose^2), family = binomial(link = logit), data = Insect)
summary(Model2)##
## Call:
## glm(formula = cbind(n_deaths, n_subjects - n_deaths) ~ dose +
## I(dose^2), family = binomial(link = logit), data = Insect)
##
## Coefficients:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -2.488172 9.867720 -0.252 0.801
## dose -0.150005 0.329179 -0.456 0.649
## I(dose^2) 0.003187 0.002727 1.169 0.243
##
## (Dispersion parameter for binomial family taken to be 1)
##
## Null deviance: 268.2683 on 7 degrees of freedom
## Residual deviance: 3.1836 on 5 degrees of freedom
## AIC: 37.962
##
## Number of Fisher Scoring iterations: 4## Analysis of Deviance Table
##
## Model 1: cbind(n_deaths, n_subjects - n_deaths) ~ dose
## Model 2: cbind(n_deaths, n_subjects - n_deaths) ~ dose + I(dose^2)
## Resid. Df Resid. Dev Df Deviance
## 1 6 4.6155
## 2 5 3.1836 1 1.4319## [1] 0.2317644两个模型比较的结果表明,无证据反对零假设(只用线性关系拟合数据是合理的),也就是说增加剂量的平方这一新的解释变量并不能提升模型对数据的拟合程度。仔细观察模型2的输出结果中,可以发现 I(dose^2) 项的 Wald 检验结果是 p = 0.24,十分接近似然比检验的结果。因为它们两者是渐近相同的 (asymptotically equivalent)。
36.5.2 低出生体重数据
- 读入该数据,试分析数据中和低出生体重可能相关的变量:
## # A tibble: 6 × 11
## id low age lwt race smoke ptl ht ui ftv bwt
## <dbl> <dbl> <dbl> <dbl> <dbl+lbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 85 0 19 182 2 [black] 0 0 0 1 0 2523
## 2 86 0 33 155 3 [other] 0 0 0 0 3 2551
## 3 87 0 20 105 1 [white] 1 0 0 0 1 2557
## 4 88 0 21 108 1 [white] 1 0 0 1 2 2594
## 5 89 0 18 107 1 [white] 1 0 0 1 0 2600
## 6 91 0 21 124 3 [other] 0 0 0 0 0 2622- 拟合一个这样的逻辑回归模型:结果变量使用低出生体重与否
low,解释变量使用母亲最后一次月经时体重lwt(磅)。
##
## Call:
## glm(formula = low ~ lwt, family = binomial(link = logit), data = lbw)
##
## Coefficients:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) 0.995763 0.785243 1.268 0.2048
## lwt -0.014037 0.006169 -2.276 0.0229 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## (Dispersion parameter for binomial family taken to be 1)
##
## Null deviance: 234.67 on 188 degrees of freedom
## Residual deviance: 228.71 on 187 degrees of freedom
## AIC: 232.71
##
## Number of Fisher Scoring iterations: 4##
## Logistic regression predicting low
##
## OR(95%CI) P(Wald's test) P(LR-test)
## lwt (cont. var.) 0.99 (0.97,1) 0.023 0.015
##
## Log-likelihood = -114.354
## No. of observations = 189
## AIC value = 232.7081- 利用
lowess平滑曲线作图,评价在 logit 单位上,lwt和low之间是否可以认为是线性关系。
pi <- M$fitted.values
# with(lbw, scatter.smooth(lwt, pi, pch = 20, span = 0.4, lpars =
# list(col = "blue", lwd = 3, lty = 1), col=rgb(0,0,0,0.004),
# xlab = "Mother's weight at last menstural period (lbs)",
# ylab = "Logit(probability) of being low birth weight",
# frame = FALSE))
plot(lbw$lwt, lbw$low, main="Lowess smoother plot\n of the prob of having a low brith weight baby",
xlab = "Weight at at last menstural period (lbs)",
ylab = "Probability")
lines(lowess(lbw$lwt, lbw$low, f = 0.7), col=2, lwd = 2)
points(lbw$lwt, pi)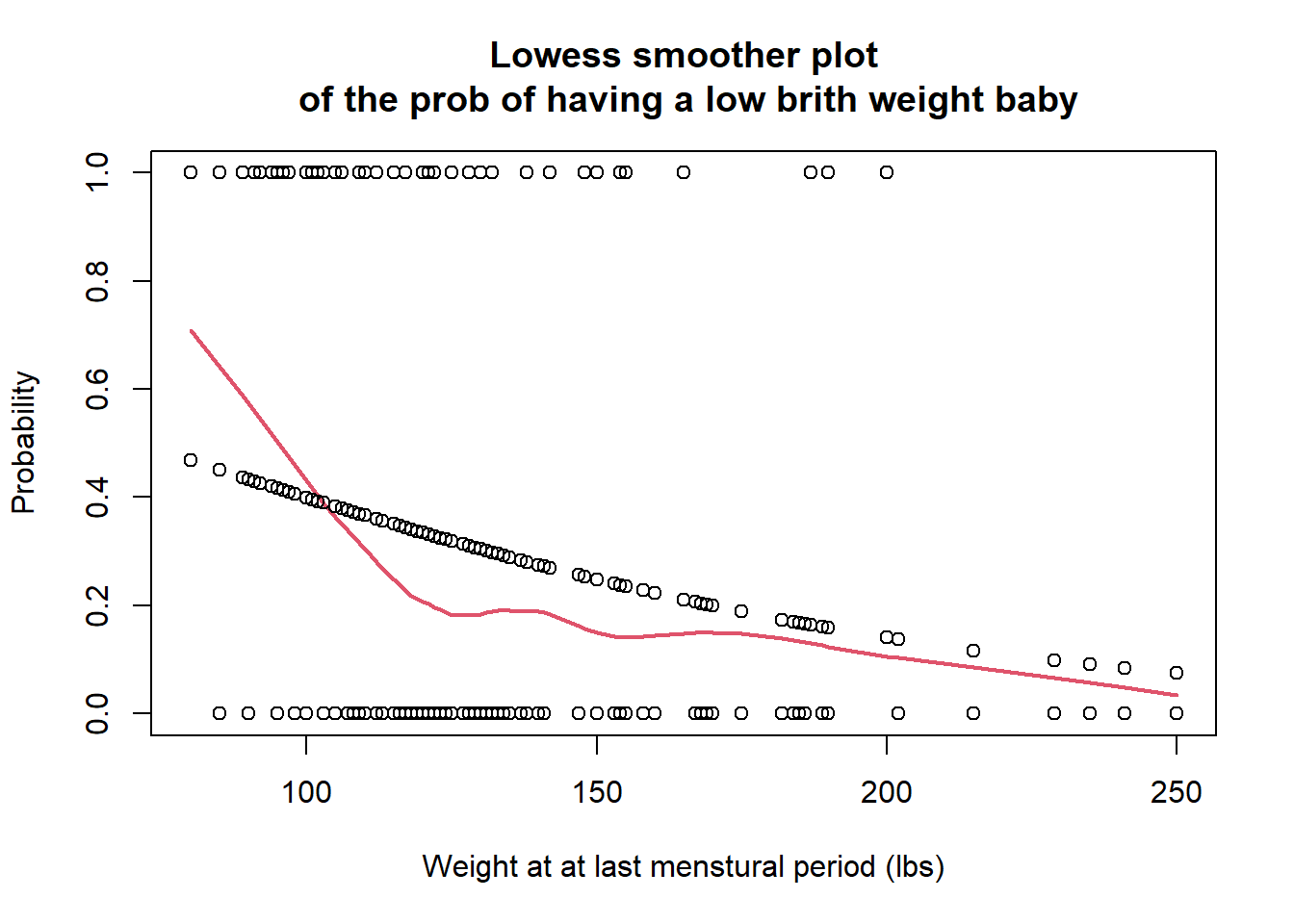
图 36.1: The loess plot of the observed proportion with low birth weight against mother’s weight at last menstural period. Span = 0.6
Lowess 平滑曲线提示模型的拟合值(fitted.values)有一些变动,由于样本采样方法等原因,这是无法避免的。但是总体来说,拟合值和平滑曲线基本在同一个步调上,从该图来看,认为母亲的最后一次月经时体重和是否生下低出生体重儿的概率的logit 之间的关系是线性的应该不是太大的问题。